








细胞自噬在维持健康和治疗疾病中的应用
 供稿:市场部
供稿:市场部 发布时间:2023-07-25
发布时间:2023-07-25 浏览量:908次
浏览量:908次
Foreword
自噬是一种细胞内部的机制,通过将细胞内成分包裹起来并运输到溶酶体进行降解,实现细胞的自我调节和清除有害物质。1963年,Christian de Duve首次提出了自噬的概念。1990年,Yoshinori Ohsumi通过酿酒酵母实验直观展现了自噬现象,并揭示了酵母和人的细胞中的自噬机制。2003年,自噬关键基因被命名为ATG基因,相关研究逐渐增加。2016年,Yoshinori Ohsumi获得诺贝尔医学或生理学奖,自噬研究进一步受到关注,成为生物学、医学、植物学和微生物学的热门研究领域。自噬的发现对于细胞调节和药物开发具有重要意义。

巨自噬(macroautophagy):通过形成具有双层膜结构的自噬体包裹胞内物质,最终自噬体与溶酶体融合。
微自噬(microautophagy):通过溶酶体或液泡表面的形状直接吞没特定的细胞器。
分子伴侣介导的自噬(chaperone-mediated autophagy,CMA):具有KEFRQ样基序的蛋白在HSP70伴侣的帮助下,通过LAMP-2A转运体转运到溶酶体。
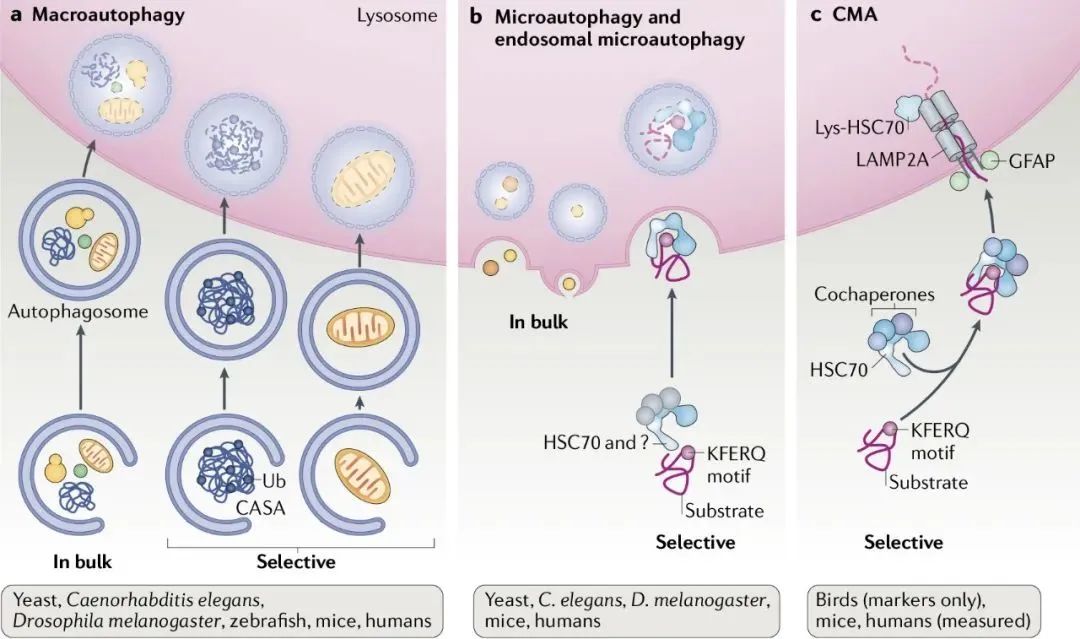
图源[1]:https://www.nature.com/articles/s41580-018-0001-6
自噬可防止细胞损伤,促进细胞在营养缺乏的情况下存活,并对细胞毒性刺激作出反应。自噬包括生理条件下的基础型自噬和应激条件下的诱导型自噬。基础自噬是一种在大多数细胞中持续发生而水平相对较低的细胞自噬过程,对细胞内物质的更新及内稳态的维持具有不可或缺作用;诱导自噬则属于一种应激状态,其发生程度明显强烈,在细胞应对外界恶劣环境维持生存中发挥重要功能。
自噬过程及核心蛋白
自噬过程主要分为自噬诱导阶段、前体成核阶段、延伸阶段、自噬体成熟阶段及自噬体与溶酶体融合阶段。
01 自噬诱导阶段(Induction)
02 前体成核阶段(Nucleation)
03 延伸阶段(Elongation)
自噬发生过程中有两组类泛素化修饰过程, 分别发生在Atg5-Atg12-Atg16连接系统和Atg8/LC3连接系统中, 用于隔离膜的延长和自噬泡的形成。
在Atg5-Atg12-Atg16连接系统中,Atg12首先由类E1泛素活化酶Atg7活化。Atg12传递给类E2泛素转移酶Atg10,与Atg10形成硫酯键。最后,Atg12与Atg5共价结合,形成Atg12-Atg5复合物。这个复合物在自噬发生时与Atg16结合形成Atg12-Atg5-Atg16。
LC3是酵母Atg8分子在哺乳动物中的同源物之一, Atg8的其他同源物还有GABARAP和GATE-16等。Atg8同源分子相比, LC3是被用作自噬发生的一个标志蛋白。LC3合成后,被AGT4B切割成LC3-I,自噬发生时,LC3-I被ATG7活化并与其形成硫酯键,然后传递给ATG3,最终在ATG5-ATG12-ATG16复合物的作用下,形成具有膜结合能力的LC3-II。参与自噬体的衔接。
04 自噬体成熟阶段与自噬体与溶酶体融合阶段
自噬体扩张和封闭过程包括自噬体外膜上ATG蛋白的清除,招募溶酶体传递蛋白和介导融合的蛋白。自噬体与溶酶体融合形成自噬溶酶体,自噬体内膜和内容物被溶酶体内的脂酶和蛋白酶降解为小分子,再利用。细胞骨架成分、运动蛋白、栓系因子、磷脂和SNAREs复合物在确保融合过程中起关键作用。
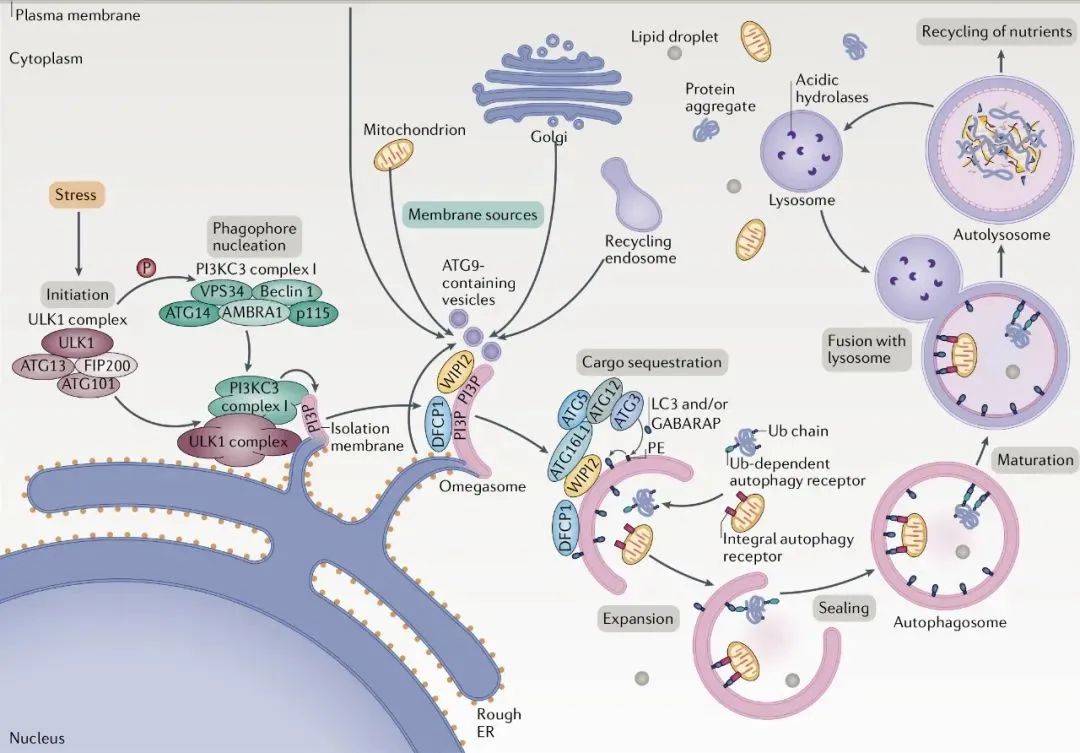
以下主要阐述起始及吞噬泡成核阶段和细胞吞噬泡延伸阶段参与的核心蛋白
1 ULK1蛋白激酶复合体中的ATG1,其功能是丝氨酸/苏氨酸激酶;通过自噬机制的磷酸化组分启动自噬;被AMPK或者mTORC1磷酸化。
2 VPS34:其功能是促进行P13KC3-C1复合体的形成,ULK1人磷酸微位点,稳定ULK1复合物;
3 Atg6/Beclin1:其功能是促进PI3KC-C1复合体的形成,调节脂质激酶VPS34;ULK1或AMPK磷酸化位点,促进自噬。
4 Atg9/mAtg9:Atg9是迄今发现的唯一一个编码跨膜蛋白的Atg基因, 可能通过影响膜泡运输对自噬发生起调控作用,其功能是将膜蛋白组分输送到吞噬泡。
5 Atg5-Atg12-Atg16:ATG5直接结合膜,这种膜结合受ATG12的负调节,但被ATG16激活。ATG12-ATG5-ATG16复合物的膜结合是有效促进ATG8脂化(LC3-I转化为LC3-II)所必需的。
6 ATG8/LC3:以LC3-Ⅰ和LC3-Ⅱ两种形式存在;参与自噬体膜的形成,与自噬体膜表面的PE结合,可作为自噬体的标记分子。
自噬与疾病研究
许多疾病,诸如癌症、神经系统疾病、炎症与免疫反应、发育和衰老等都与细胞自噬密切相关。科学研究者也要积极探索自噬与这些疾病之间的联系,努力揭示自噬调控分子机制,通过抑制或促进调节自噬过程,从而为各种疾病治疗提供新的方向和可能性。
自噬与神经系统疾病
临床研究发现,在许多神经退行性疾病中, 病变区域常有大量的泛素化蛋白质聚合物,这些多因错误折叠而被泛素化的蛋白质聚合物正是引起神经退行性疾病的重要原因之一。
小鼠中特异性敲除神经元中的自噬基因ATG5或ATG7,会抑制自噬发生的水平,同时伴随着大量泛素化蛋白质聚合物的积累,并最终引发神经退行性疾病[3-4]。在小鼠和果蝇模型中,用Rapamycin或其类似物抑制mTOR的活性促进自噬发生,可以缓解神经退行性疾病的症状[5-6]。
由此可见,诱导自噬可能成为一种治疗神经系统疾病的治疗方法。其次是包括帕金森症在内的一些神经退行性疾病。
自噬与肿瘤发生的联系是当今自噬研究的一个重要热点问题。自噬功能正常, 对肿瘤起抑制作用, 而敲除自噬基因将引发肿瘤的形成。Beclin1其实就是一个肿瘤抑制基因,干扰Beclin1的表达使自噬受到抑制, 会增加肿瘤发生的可能[7]。在胸腺癌细胞系中过表达Beclin1, 可以减缓癌细胞的生长速度并减少其致癌性[8]。UVRAG能够结合Beclin1/Vps34复合物促进自噬, 而UVRAG同时有抑制肿瘤的功能[9]。Bif-1可以通过UVRAG结合Beclin1增强自噬, 敲除Bif-1, 显著加快了肿瘤的生长[10]。
自噬受阻可能导致细胞失去生长调节及启动程序性死亡的能力, 同时还可能阻碍细胞分解代谢, 使p62、受损的线粒体、蛋白质聚合物、ROS等有害物质积累, 并进一步损伤基因组DNA的稳定性, 致使原癌基因激活, 而以上这些最终都将导致肿瘤的发生。
因此,在不同的环境因素或稳定状态下,自噬既可以阻止也可以促进肿瘤的发生。
自噬与免疫
自噬是至今已发现的唯一可以降解细胞器和较大蛋白质聚集物的细胞降解过程,因此也被预测可能参与降解外来微生物的过程[11]。许多研究发现,一些自噬蛋白甚至整个自噬过程都直接参与免疫和炎症反应[12]。巨噬细胞中,干扰素IFN-γ可以诱导自噬来抵御分枝杆菌等病原菌,而且在已感染的巨噬细胞中, 用IFN-γ预处理可以引起自噬泡吞噬含有病原菌的膜泡并与溶酶体融合,增强其对胞内细菌的杀伤力。血管平滑肌细胞中, 肿瘤坏死因子TNF-α通过激活JNK通路和抑制Akt,上调LC3和Beclin1的表达促进自噬[13]。
由此可见,自噬与细胞免疫的发生必然存在密切的联系。
【参考文献】
1.Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. 2018 Jun;19(6):365-381.
2.Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Biol. 2018 Jun;19(6):349-364.
3.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 2006; 441(7095): 885-9.
4.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, etal. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006; 441(7095): 880-4.
5.Sarkar S, Perlstein EO, Imarisio S, Pineau S, Cordenier A, Maglathlin RL, et al. Small molecules enhance autophagy and reduce toxicity in Huntington’s disease models. Nat Chem Biol 2007; 3(6): 331-8.
6.Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, et al. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of.
7.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA 2003; 100(25): 15077-82.
8.Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402(6762): 672-6.
9.Liang C, Feng P, Ku B, Dotan I, Canaani D, Oh BH, et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nat Cell Biol 2006; 8(7): 688-99.
10.Takahashi Y, Coppola D, Matsushita N, Cualing HD, Sun M, Sato Y, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nat Cell Biol 2007; 9(10): 1142-51.
11.Kraft C, Peter M, Hofmann K. Selective autophagy: Ubiquitinmediated recognition and beyond. Nat Cell Biol 2010; 12(9): 836-41.
12.Jia G, Cheng G, Gangahar DM, Agrawal DK. Insulin-like growth factor-1 and TNF-alpha regulate autophagy through c-jun N-terminal kinase and Akt pathways in human atherosclerotic vascular smooth cells. Immunol Cell Biol 2006; 84(5): 448-54.
13.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages.Cell 2004; 119(6): 753-66

 返回列表
返回列表
在线咨询
Online consultation
-
 在线咨询
在线咨询
-
 技术支持
技术支持

关注微信公众号

